Một phương pháp mới giúp giải mã sinh học tiến hoá
Một công cụ tin sinh học (Bioinformatic) mới để so sánh dữ liệu gen đã được phát triển bởi một nhóm nghiên cứu của đại học tổng hợp và đại học Y khoa Vienna của Áo, cùng với các cộng sự từ Úc và Canada. Chương trình máy tính này có tên là “ModelFinder” sử dụng một thuật toán rất hiệu quả và cho phép một cái nhìn mới về tiến hoá. Kết quả được xuất bản ngày 8/5/2017 trên tạp chí có uy tín thế giới, Nature Methods.
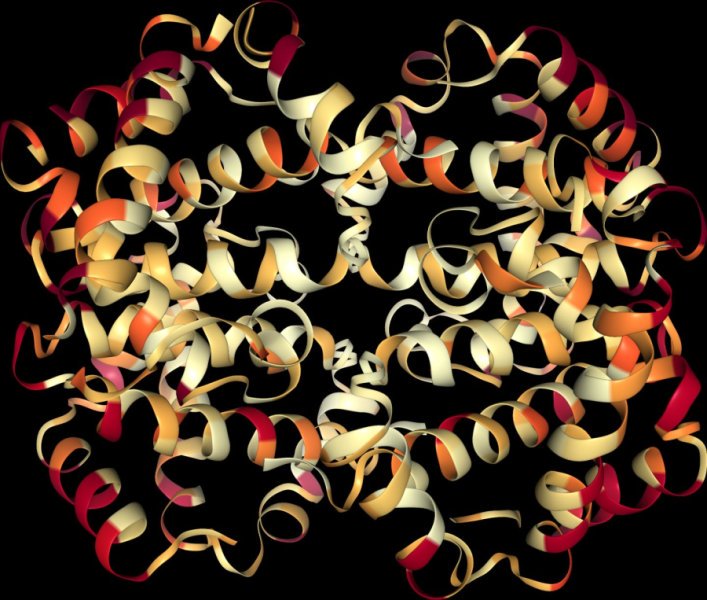
Cấu trúc không gian ba chiều của chuỗi protein huyết sắc tố (Hemoglobin) của loài người, trong đó các đoạn có tốc độ tiến hoá nhanh (màu đỏ) và chậm (màu vàng) được tính toán bởi ModelFinder (Bản quyền: Bùi Quang Minh, Đại học tổng hợp Vienna, Áo).
Tìm hiểu về tiến hoá là một trong các nền tảng cơ bản của sinh học – tiến hoá chính là lời giải đáp duy nhất về đa dạng sinh học trên trái đất.
Dựa trên sự tiến hoá của các chuỗi protein, các nhà khoa học có thể giải thích được sự hình thành các loài và các chức năng thông qua biến đổi gen, làm thế nào để tạo ra chức năng mới với các Enzim, hoặc là loài người có quan hệ tiến hoá như thế nào với khỉ và tinh tinh.
Một phương pháp phổ biến để nghiên cứu tiến hoá là bằng cách so sánh dữ liệu hệ gen sử dụng các công cụ tin sinh học. Từ đó các nhà khoa học có thể so sánh các chuỗi protein, được hình thành từ sự kết hợp của 20 loại Axit amin.
Cho đến nay, các công cụ tin sinh học này đều giả thiết rằng tố độ tiến hoá của các đoạn protein xuất phát từ một mô hình phân phối xác suất thống kê có một biến số duy nhất.
Bùi Quang Minh, đồng tác giả chính của nghiên cứu này, giải thích: “Tuy nhiên giả thiết này không đúng với thực tế, và điều đó có thể dẫn đến nhiều kết quả nghiên cứu trong vòng hai thập kỷ trở lại đây bị sai lệch”.
Một thuật toán mới về sự tiến hoá của các protein
Giáo sư Arndt von Haeseler, trưởng nhóm nghiên cứu của viện Max F. Perutz và giáo sư Lars Jermiin từ trường đại học quốc gia Úc cùng cộng sự đã tìm ra một phương pháp mới về tốc độ tiến hoá sử dụng mô hình tin sinh học.
Rất nhiều chuyên gia đều đồng ý rằng phương pháp phổ biến hiện nay có thể không nắm bắt được độ phức tạp của sự tiến hoá các protein. Tuy nhiên, mức độ tính toán của các mô hình thực tế hơn đều quá phức tạp. Giáo sư Lars Jermiin nói: “Chúng tôi đã giải quyết được vấn đề này bằng một thuật toán rất hiệu quả, điều đó cho phép chúng ta có hiểu biết sâu sắc hơn về sự tiến hoá của các protein. Công cụ mới này sẽ có tác động lớn đến nhiều lĩnh vực nghiên cứu, ví dụ sự tiến hoá của các mầm bệnh vi sinh vật hoặc sự phát tán của các loài gây hại.”
Chương trình máy tính “ModelFinder” sẽ đưa ra được đánh giá chính xác hơn về các quá trình tiến hoá. Điều này sẽ giúp chúng ta tiến thêm một bước để làm sáng tỏ các bí mật của tạo hoá và sự sống trên trái đất.
Bài báo xuất bản trên Nature Methods:
“ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates”. Subha Kalyaanamoorthy, Bui Quang Minh, Thomas KF Wong, Arndt von Haeseler, Lars S Jermiin. Nature Methods (published online 8 May 2017)
http://dx.doi.org/10.1038/nmeth.4285
Nguồn dịch từ ScienceDaily
http://sciencedaily.com/releases/2017/05/170509093628.htm
Người dịch: TS. Bùi Quang Minh, đồng tác giả chính của nghiên cứu, từng học tập và nghiên cứu tại trường Đại học Công nghệ, Đại học Quốc gia Việt nam, Hà nội. TS. Bùi Quang Minh sau đó sang Đức và Áo để học sau đại học và tiến sỹ, trước khi trở thành chuyên viên nghiên cứu của ĐH Tổng hợp Vienna, Cộng hoà Áo.
